Direktoxidationsverfahren zur Herstellung von für Epoxiden

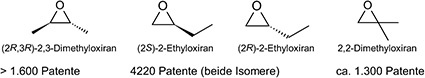

Als „Epoxide" schlechthin werden meist nur zwei Verbindungen bezeichnet: Ethylenoxid und Propylenoxid. Von den höhermolekularen Epoxiden ist nur noch 1,2-Butylenoxid in technischen Mengen zu haben. Produkte wie etwa 2,3-Butylenoxid oder 4-Methyl-pentylen-1,2-oxid sind auf dem Markt kaum verfügbar und finden daher auch keinen Eingang in technische Anwendungen.
Dabei eröffnen sie beinahe unerschöpfliche Möglichkeiten, die Eigenschaften von Produkten wie Schmierstoffen, PU-Schäumen und Beschichtungsmaterialien zu verändern oder ganz neue Eigenschaften einzuführen. Die hohe Zahl diesbezüglicher Patentanmeldungen zeigt das bestehende Interesse.
Epoxide haben ein Marktvolumen von knapp 30 Mio. t. Während die mengenmäßig größte Anwendung der Epoxide die Umsetzung zu Glykolen ist, ist ein weiteres wichtiges und technisch anspruchsvolleres Einsatzgebiet die Verwendung als Monomer für die Herstellung von Polyurethanen und Polyethern.
Epoxide in Polyurethanen
Bei keinem anderen technischen Polymer kann eine solche Variation der Eigenschaften allein über die chemische Struktur eingeführt werden wie bei Polyurethan. Polyurethane können für so verschiedene Endprodukte wie Weichschäume oder harte Kunststoffbauteile verwendet werden. Im Gegensatz zu z. B. PVC wird die Variabilität nicht in erster Linie durch den Einsatz von Füllstoffen erreicht. Polyurethane setzen sich zusammen aus einer Isocyanatkomponente und einem Polyether-Polyol, welches seinerseits aus Epoxiden hergestellt wird.
Der Gewichtsanteil des Polyethers am Polyurethan liegt in jedem Fall über 50 % und erreicht bei Weichschäumen mit besonders langkettigen Polyether-Polyolen Werte von über 97 %. Zwar beeinflussen auch das gewählte Isocyanat und die Funktionalität des Polykondensationsstarters die endgültigen Eigenschaften des fertigen Polymers, aber die Chemie der Polyetherketten hat einen entscheidenden Einfluss.
Bereits in den 80er Jahren wurden etliche Modifikationen von Polyurethanen durch die Verwendung höhermolekularer Epoxide patentiert, mangels Marktverfügbarkeit wurde jedoch nur wenig davon umgesetzt. Im Folgenden sind einige Beispiele für Anwendungsmöglichkeiten aufgeführt:
Leichtere Schäume
Polyurethane können nicht nur durch hydrolytisch abgespaltenes CO2, sondern auch mit Pentan geschäumt werden, ähnlich wie Polystyrol. Ein (teilweiser) Ersatz von Ethylenoxid durch ein höhermolekulares Epoxid, wie Butylenoxid, macht die ursprünglich hydrophile Polymerkette lipophiler, so dass sich deutlich mehr Pentan im Vorgelat lösen lässt. Das Ergebnis: Ein Hartschaum mit vermindertem spezifischen Gewicht.
Höhere Wasserbeständigkeit
Die Verminderung der Wasserlöslichkeit der Polyetherketten führt nicht nur zu einer besseren Löslichkeit fettähnlicher Stoffe, sondern auch zu einer verminderten Angreifbarkeit durch Wasser. Höhermolekulare Epoxide können als Comonomere Polyurethanbauteilen, die Wasser oder Dampf unter aggressiven Bedingungen ausgesetzt sind, ein entscheidendes Plus an Beständigkeit verleihen.
Wirkstoffpflaster
Hormone, Nikotin, Schmerzmittel und andere Wirkstoffe können über ein Pflaster durch die Haut verabreicht werden. Diese Darreichungsform ist einfach anzuwenden und für eine Reihe von Wirkstoffen die bevorzugte galenische Form. Um den Wirkstoff in möglichst wenig Pflastermaterial einzubringen und die Freisetzung zu kontrollieren, muss das wirkstofftragende Polster chemisch genau auf das darin deponierte Molekül abgestimmt werden. Das kann z. B. dadurch geschehen, dass das Polyurethan durch die Verwendung höhermolekularer Epoxide deutlich lipophiler gestaltet wird.
Dichtungsschutz in Kältemaschinen
Bei der Verwendung von Ammoniak als Kältemittel mit kohlenwasserstoffbasierten Schmiermitteln neigen die Dichtungen zum Schrumpfen und Verhärten. Dies führt zu Kältemittelverlust und erfordert es letztlich, die Anlage zu entleeren und zu warten. Ein kleiner Zusatz von Poly-(1,4-Butylenoxid), eines relativ öllöslichen Polyethers, zum Schmiermittel erhält dagegen das Volumen und die Flexibilität der Dichtungen.
Was macht diese Epoxide so rar?
Wenn also höhermolekulare Epoxide eine solche Anwendungsvielfalt haben, warum kann man sie dann nicht kaufen? Die Antwort liegt im höheren Herstellaufwand, der bislang die Entwicklung eines Marktes für diese Verbindungen verhindert hat.
Ethylenoxid und Propylenoxid lassen sich in einstufigen, seit Jahren gut etablierten Prozessen herstellen. Epoxide mit einer höheren Kohlenstoffzahl zu produzieren, ist dagegen deutlich aufwändiger: Bereits Butylenoxid muss durch einen zweistufigen Prozess hergestellt werden, der Oxidation von Butadien zu Vinyloxiran und dessen katalytische Hydrierung zu 1,2-Butylenoxid. Noch höhermolekulare Epoxide lassen sich durch ähnliche, manchmal auch noch längere Reaktionssequenzen herstellen. Eine Direktoxidation von Olefinen mit einer höheren Kohlenstoffzahl als 3 ist nur für einzelne Olefine möglich.
Direktoxidation von Olefinen
Durch ein am Leibnitz-Institut für Troposphärenforschung in Leipzig entwickeltes Direktoxidationsverfahren mit Ozon und NO2 ist es jetzt möglich, zahlreiche Monoolefine mit Kohlenstoffzahlen von C2 bis C6 in einer Gasphasenoxidation zum entsprechenden Epoxid umzusetzen. Die Reaktion läuft bei mittleren Reaktionstemperaturen (zwischen 300 und 400°C) und kommt ohne Katalysator aus. Die Selektivität der Oxidation ist sehr hoch, es fallen nur geringe Mengen der jeweiligen Aldehyde als Nebenprodukt an. Die günstigen Rahmenbedingungen ermöglichen es, aus einer Produktionsanlage verschiedene höhermolekulare, verzweigte und/oder funktionalisierte Epoxide zu erhalten, und zwar ohne Umrüstung.
Dieses Herstellverfahren ist gegenüber den bisher zur Verfügung stehenden Synthesewegen eine ausgesprochene Abkürzung und eröffnet den Weg nicht nur zu modifizierten Polymeren. Auch die entsprechenden Glykole werden so leichter zugänglich, und auch für die Synthese kleiner Moleküle können die höhermolekularen, sehr reaktiven Epoxide interessante Ausgangsmaterialien sein.
Anbieter
Dr. Arnold UnternehmensberatungHinterm Ließ 13
88481 Balzheim
Deutschland








Meist gelesen

Chemiekonjunktur – China auf der Überholspur
Im Jahr 2024 stieg der Anteil Chinas an den globalen Chemieinvestitionen auf rund 45 %. Doch die goldenen Jahre des chinesischen Wirtschaftswunders sind vorbei.

Radikaler Schnitt für gesundes Wachstum – CHT stellt sich neu auf
Die CHT Group gehört zu den Hidden Champions der deutschen Spezialchemie.

Vorsprung durch Forschung
Wacker baut seine Forschungsaktivitäten aus. Der Münchner Chemiekonzern hat einen zweistelligen Millionenbetrag in ein neues Biotechnology Center in der bayerischen Landeshauptstadt investiert.

Verluste, die keiner sieht
In CCUS-Projekten der chemischen Industrie wird die präzise CO₂-Messung zur Schlüsseltechnologie - Coriolis-Systeme spielen dabei eine zentrale Rolle.

Schlüsselrohstoff für Industrie und Gesellschaft
Hochreines Salz besitzt für Europa strategische Bedeutung – doch der Markt ist konzentriert.