Beschleunigte Wirkstoffforschung
Im Januar gaben Schrödinger, ein Anbieter integrierter Software-Lösungen, und das Life-Sciences-Unternehmen Bayer eine auf 5 Jahre angelegte Technologieallianz zur Entwicklung einer umfassenden De-novo-Designlösung mit dem Ziel bekannt, die Entdeckung innovativer, qualitativ hochwertiger Medikamente zu beschleunigen.
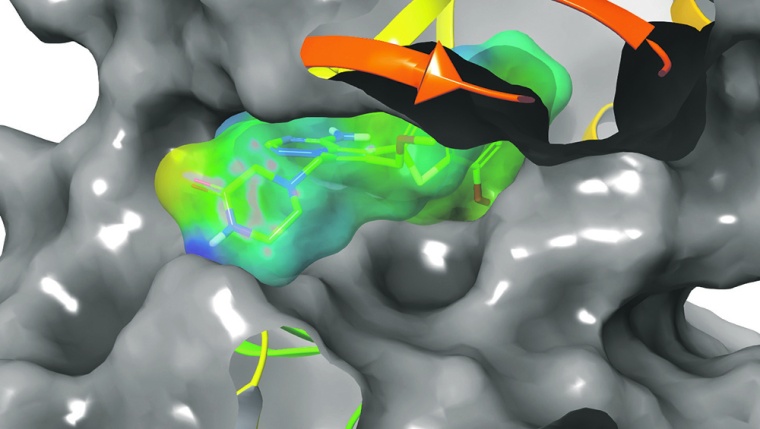
Die beiden Unternehmen gehen davon aus, dass die Technologie in der Lage sein wird, eine sehr große Anzahl synthetisch herstellbarer, virtueller Verbindungen zu erfassen, in silico – also in Computermodellen – zu screenen und zu bewerten, um die Identifizierung und Optimierung potenzieller neuer therapeutischer Kandidaten zu unterstützen. Jörg Weiser, Managing Director von Schrödinger, und Alexander Hillisch, Head of Computational Molecular Design bei Bayer, erläutern CHEManager die Hintergründe der Kooperation und geben einen Einblick in aktuelle und zukünftige Möglichkeiten der Wirkstoffforschung. Die Fragen stellte Ralf Kempf.
CHEManager: Herr Weiser, Herr Hillisch, um was geht es bei der Anfang 2020 bekannt gegebenen Kooperation zwischen Schrödinger und Bayer?
Jörg Weiser: Die fünfjährige Allianz unserer Unternehmen hat die Entwicklung einer eigenen, umfassenden De-novo-Wirkstoffdesign-Software zum Ziel. Die Technologie soll in der Lage sein, Milliarden synthetisch herstellbarer, virtueller Verbindungen zu spezifizieren, auszusieben und zu bewerten, um so die Identifizierung und Optimierung potenzieller neuer Arzneimittelkandidaten zu unterstützen.
Die De-novo-Designplattform kombiniert Schrödingers Molekular-Design-Technologie, die auf physikbasierter Modellierung, maschinellem Lernen und Enterprise-Informatik (LiveDesign) beruht, mit Bayer-eigenen In-silico-Modellen, die auf dem enormen Wissens- und Datenschatz von Bayer fußen und Vorhersagen zu Absorption, Verteilung, Metabolismus, Ausscheidung, Toxizität (ADMET) und chemischer Synthetisierbarkeit von Verbindungen ermöglichen. Die gemeinsam zu entwickelnde Lösung soll in Zukunft neue und schnellere Wege für die Wirkstoffforschung erschließen.
Was genau ist eine De-novo-Design-Plattform?
Alexander Hillisch: Das ist eine Software-Plattform, die in der Lage ist, potenzielle neue Wirkstoffe computerbasiert vorzuschlagen. In der Wirkstoffentwicklung ist De-novo-Design die aktuellste Entwicklungsstufe. Es unterscheidet sich von älteren Methoden darin, dass dabei komplett neue Wirkstoffe entworfen werden und gleichzeitig wichtige Eigenschaften wie Bioverfügbarkeit und Löslichkeit, sowie die Herstellbarkeit der Verbindungen im Labor mitberücksichtigt werden. Wir nennen dies holistische Ansätze. Trotz der Tatsache, dass physikbasierte Computersimulationen und künstliche Intelligenz den Kern dieser Ansätze darstellen, erfordern diese tiefgreifendes menschliches Wissen in der Bedienung und Auswertung der Computerprogramme, sowie der Herstellung und biologischen Testung von Verbindungen im Labor.
Wie kam es zu der Zusammenarbeit der beiden Unternehmen? Gab es schon frühere gemeinsame Projekte?
J. Weiser: Bayer und Schrödinger arbeiten seit 2003 kontinuierlich zusammen. Aus einer Software-Kundenbeziehung entwickelte sich mit der Zeit eine Partnerschaft, bei der einzelne kleinere Projekte hinsichtlich der Verbesserung von computerbasierten Wirkstoffdesign-Methoden durchgeführt wurden. Bereits 2013 fand ein gemeinsames Treffen in New York statt, bei dem man über die Möglichkeiten zur vertieften Zusammenarbeit an einer gemeinsamen zu entwickelnden Plattform sprach. Diese Ideen werden jetzt konkret realisiert.
Warum die dreifache Integration von Physik, Machine Learning und Enterprise-Informatik?
J. Weiser: Das Gravitationszentrum erfolgreicher Medikamentenforschung ist das Design von Molekülen mit den gewünschten Eigenschaften. Will man diese auf dem Niveau experimenteller Ergebnisse berechnen, geht kein Weg an physikalischen Methoden vorbei. Man braucht aber auch möglichst viele Ausgangsmoleküle – Millionen bis Milliarden – um eine gute Chance zu haben, in diesem chemischen Raum einige, wenige optimale Moleküle herauszufischen. Ideengenerierung kann sowohl mit Hilfe von Machine Learning als auch mit Chemieinformatik erfolgen. Zudem können mit diesen Methoden anfängliche Filterrechnungen erfolgen. Alle Moleküle, alle Rechnungen und auch alle Experimente müssen dann zusammengeführt und ausgewertet werden können, um die besten Entscheidungen zu treffen. Dazu benötigt man ein Enterprise-Informatik-System, das sowohl virtuelle als auch experimentelle Daten erfasst, analysiert und für Entscheidungsfindungen vernünftig aufbereitet.
„Das Gravitationszentrum erfolgreicher Medikamentenforschung ist das Design von Molekülen mit den gewünschten Eigenschaften.“
- Jörg Weiser, Schrödinger
Wie beeinflusst die digitale Transformation die Entwicklung neuer Medikamente?
A. Hillisch: Aufgrund der Komplexität des menschlichen Körpers und der Biologie ist der bisherige Wirkstofffindungs- und Entwicklungsprozess sehr von ‚trial and error‘ dominiert. Es müssen an vielen Zielproteinen, den sogenannten Targets, sehr viele Verbindungen getestet werden, um zu einem neuen Wirkstoff zu gelangen.
Digitale Methoden bieten hier ungeahnte Möglichkeiten zur Verbesserung der Effizienz des Wirkstofffindungs- und Entwicklungsprozesses. Angefangen bei der Auswahl von Targets über Datenanalyse von genomischen Zusammenhängen sowie bei der Auffindung und Optimierung von Leitstrukturen, zum Beispiel durch virtuelles Screening oder De-novo-Design. Auch bei der Herstellung von Wirkstoffen kommt maschinelles Lernen zur Anwendung, indem jahrzehntelanges Synthese-Know-how computerbasiert ausgewertet und angewendet wird. Besonders bei der aufwendigen klinischen Entwicklung sind die Chancen riesig. Das Design von klinischen Studien kann durch In-silico-Analysen von genomischen Zusammenhängen optimiert werden und bei ihrer Durchführung könnten künstliche Intelligenz und Sensorik die Therapietreue erhöhen und damit schneller zu Ergebnissen führen.
„Digitale Methoden bieten ungeahnte Möglichkeiten zur Verbesserung der Effizienz des Wirkstofffindungs- und Entwicklungsprozesses.“
- Alexander Hillisch, Bayer
Für die Forschung liegt der größte Vorteil digitaler Ansätze aber darin, dass In-silico-Analysen möglich sind, die experimentell gar nicht durchführbar wären und damit eine neue Qualität bieten. Digitale Methoden werden in der Pharmaforschung aber immer Experimente triggern und eng mit diesen verknüpft sein.
Wie verändert die zunehmende Digitalisierung die Tätigkeiten von Forscherinnen und Forschern?
A. Hillisch: Der Computer macht, was er am besten kann – er bietet enormes Rechenvolumen. Das ermöglicht Forscherinnen und Forschern sich ganz auf Design, Prozessoptimierung und kontextuelle Entscheidungen zu fokussieren. Wir werden zukünftig mehr Menschen in der Forschung in diesen Bereichen benötigen. Sie behalten den Gesamtüberblick und sorgen für die Feinjustierung einer Plattform für ein konkretes Projekt.
Wie seit jeher in der Forschung werden sich die Berufsbilder stetig ändern. Was bleibt, ist die Notwendigkeit zur menschlichen Kooperation, denn die kontextuellen Entscheidungen werden von Menschen gefällt werden, nicht von Computern, nicht von Maschinen.
Was macht eine digitale Plattform attraktiv?
J. Weiser: Die Echtzeitanalyse zusammengeführter virtueller und experimenteller Ergebnisse – das gab es noch nie. Außerdem ermöglichen digitale Plattformen viel engere Kooperation von Forschern und Forschergruppen – das hat die wissenschaftliche Gemeinschaft weltweit während der Covid-19-Pandemie eindrucksvoll unter Beweis gestellt. Die Vernetzung bedeutet auch die Aufhebung von Datensilos und die Integration von Partikularwissen. Außerdem können wir bereits heute Entscheidungen in bisher unbekanntem Maße auf Datenanalysen stützen.
Zudem lernen wir mit jeder Analyse, denn durch Machine Learning wird ein Großteil der Informationen, die eine Organisation in den letzten Jahrzehnten gesammelt hat bzw. in silico mit hockakkuraten physikalischen Methoden generieren kann, in prospektiv anwendbares Wissen umgewandelt und steht als Berechnungsmethode für neue Aufgaben jederzeit zur Verfügung.
Was bedeutet das konkret für Bayer?
A. Hillisch: Wir werden mit De-novo-Ansätzen bei bestimmten Projekten in der Wirkstoffforschung schneller und mit weniger Aufwand zum Ziel kommen, also Leitstrukturen finden und Wirkstoffe optimieren. Natürlich erhoffen wir uns, so auch Aspekte verfolgen zu können, die vorher nicht möglich waren. Durch holistische Betrachtungsweisen von Milliarden von virtuellen Verbindungen sollen Eigenschaften von Verbindungen gezielt verbessert werden können – ohne Kompromisse bei anderen Eigenschaften eingehen zu müssen. Dies ist innerhalb einer eher eng begrenzten Molekülserie durchaus schwierig zu erzielen. Mit dem De-novo-Ansatz glauben wir, dies besser erreichen zu können.
Wie stellt sich Schrödinger die weitere Zukunft der Wirkstoffforschung vor?
J. Weiser: Wir gehen davon aus, dass unsere Plattform in Zukunft für das molekulare Design in Pharma, Crop, Green Biotech, Lebensmittel, Materialien usw. maßgeblich sein wird. Die Integration von physikalischen Methoden, Enterprise-Informatik und Machine Learning wird weiter voranschreiten. Die Qualität von virtuellen Daten und die Geschwindigkeit deren Generierung wird sich weiter verbessern und damit stetig relevanter für die Entscheidungsfindung in Forschungsprozessen werden. Auch die Zusammenarbeit von Forscherinnen und Forschern wird sowohl global als auch in einem Unternehmen noch enger werden. Damit steigt die Bedeutung, auf Basis von kollektiver menschlicher Intelligenz datenbasierte Entscheidungen zu fällen – das wäre dann eine schöne Aufhebung des industriellen Widerspruchs bezüglich der Kontrollverhältnisse zwischen Mensch und Maschine in eine produktive Synthese. Hegel hätte zu seinem 250. Geburtstag bestimmt seine Freude daran.
___________________________________________________________
Accelerated Drug Discovery - die englische Fassung lesen Sie hier.
___________________________________________________________






