For Increased Patient Safety

Drug Safety - To fight the alarming increase of falsified medicinal products detected, the European Parliament and the Council of the European Union released the Directive 2011/62/EU in the middle of last year. From the pharmaceutical industry's perspective, the key measures described are a requirement for safety features that will allow to validate the authenticity of drugs and the introduction of a system to verify individual medicinal packs.
As a general rule, all prescription medicines will have to bear the safety features unless they find their way onto the ‘whitelist' of exemptions. For non-prescription OTC products, it's vice-versa: They are exempted from the safety feature requirements unless they are part of the ‘blacklist' e.g. as a result of an assessment of the falsification risk.
The manufacture of active pharmaceutical ingredients (APIs) will have to comply with the principles and guidelines of good manufacturing practice (GMP) even if they are produced outside of the EU. Amongst others, this will be ensured through audits of the API manufacturers as well as by a written confirmation of the pharmaceutical manufacturer that he himself has verified compliance of his API manufacturer with GMP rules.
Concept Paper for Public Consultation
Whilst the directive already covers GMP for APIs extensively, details regarding the other topics will be determined separately by means of ‘Delegated Acts' that the EU Commission is going to enact. In preparation thereof, the commission published a concept paper in November 2011 and has invited all stakeholders to comment. The document outlines different scenarios for the individual topics including pros and cons and lists a set of consultation items to be explicitly addressed in the responses.
Presumably, the policy option to leave the product coding scheme to each individual manufacturer's choice is just mentioned for the sake of completeness as it would lead to an unmanageable coding variety in Europe.
Getting this harmonised as part of the ‘Delegated Acts' is therefore a must that needs to cover a product number and a randomised serial number at a minimum. Batch number and expiry date are two additional conceivable elements of a harmonised product coding scheme, in idea that is jointly supported by the European Federation of Pharmaceutical Industries and Associations (EFPIA) together with the European associations of community pharmacists (PGEU), full-line wholesalers (GIRP), and parallel distributors (EAEPC).
With regard to the question whether to use a linear barcode, a two-dimensional barcode or an RFID chip as data carrier, EFPIA and the other associations are equally aligned and together vote for the usage of the two-dimensional Data Matrix code.
Verification of Drugs
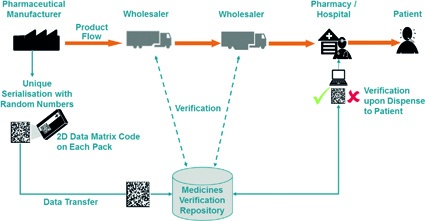
Anticipating the European law making process currently underway, EFPIA developed its ‘Point-of-Dispense' verification model that is based on the coding of each medicinal pack with product number, randomised serial number, batch ID and expiry date. This data is encoded in a Data Matrix code and printed on the pack by the manufacturer who simultaneously transfers this information to a central verification repository.
Upon dispense of the medicine in a pharmacy, the pharmacist scans the pack code and his Point-of-Sales system launches a verification request to the central repository. If the latter knows a pack of the scanned identity, it marks the pack as ‘dispensed' and the pharmacist hands it over to the patient. If the verification request fails however, the pharmacist knows that he holds a pack in hands that he must not dispense to the patient.
In 2009 / 2010, EFPIA has proven the feasibility of its verification model in a pilot project where the concept was successfully applied in day-to-day operations of selected Swedish pharmacies. Furthermore, the model is well in line with what is outlined in the commission's concept paper: The ‘checkout' of each pack in the verification repository upon dispense to the patient must be mandatory.
Directive 2011/62 already includes one provision that is important from the manufacturers' perspective: The costs of the central repository will have to be borne by them. But how could the architecture of such a system look like for an EU with currently 27 member states? And what about the accompanying organisation? Regarding the latter, the concept paper takes three alternatives into consideration:
In case of a stakeholder-governed organisation, the ‘Delegated Acts' would set out the objectives of the verification system to be achieved and define the obligations on the relevant stakeholders but leave everything else - such as the establishment and operation of the system as well as the rules for usage - to the collaborative decision of pharmaceutical companies, wholesalers, and pharmacists.
The other two alternatives for the organisational setup that the concept paper sets forth are the ‘EU Governance' by the EU Commission themselves or the European Medicines Agency (EMA) acting on their behalf and the ‘National Governance' by official national bodies.
System Architecture and Organisational Aspects
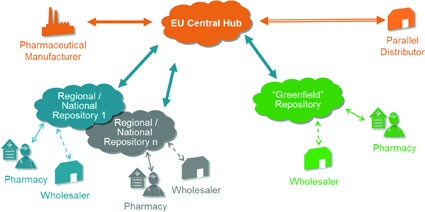
With respect to the system landscape, EFPIA together with its partners from the other stakeholder associations propose a setup that is composed of an EU Central Hub and connected national systems. The EU Central Hub serves as the entry point for pharmaceutical companies and parallel distributors to feed their pack data via a single European gateway into the different national systems. In each country, both product verification and checkout of the pack upon dispense occur against the data contained in the national repository.
For the development of the national systems, different variants can be thought of: National stakeholder organisations could decide to develop their own system e.g. in order to perfectly embed it into a pre-existing IT infrastructure in the pharmacies. The downside of this approach is the need to acquire the necessary knowhow and to bear the costs for system design and implementation all alone.
It could as well be that several countries liaise to develop and operate a joint system. And finally, the ‘Greenfield' approach is an offer where a European stakeholder organisation would develop one verification repository template that would then be parameterised to national needs so as to end up with a set of parameterised Greenfield instances that are all operated by the European organisation on behalf of the individual national stakeholder organisations.
Thismakes clear: Out of the three organisation alternatives outlined in the concept paper, it's the ‘Stakeholder Governance' model that is preferred by manufacturers, wholesalers, and pharmacists. Thereto Dr. Martin Friedrich, Head ofProduct Tracking and Authentication at Bayer Technology Services GmbH and Program Manager at EFPIA: "Having all partners at one table is key for the success and makes concerns unfounded that the pharmaceutical industry could try to enforce any distinct interests to compensate that they have to pay for the system."
It's Time to Start - Now
What are the next steps? Once having received all answers to the November public consultation paper in the end of April, the EU Commission will publish all answers and comments on their website. This will serve as an input for the further work of the commission that along several steps will finally result in the publication of the ‘Delegated Acts' in 2014.
Subsequently, the EU member states will have three years time to turn them into national law. Seemingly a long timeframe but the experience from the requirements of the Turkish Ministry of Health and the introduction of the non-serialised Data Matrix coding in France reveals the considerable amount of work that manufacturers are facing.
Thereto again Dr. Friedrich: "At first, each company needs to have a master plan that considers the existing IT infrastructure and all packaging sites delivering to the EU with their typically widespread equipment diversityas it evolved over time. The next steps are then the revision of the medium-term budget planning and the selection and assignment of capable suppliers. Those who start late will not be ready in time as there is already a competition for the short resources in the market."



most read
ISPE Good Practice Guide: Validation 4.0
The Validation 4.0 Guide provides a comprehensive approach to ensuring product quality and patient safety throughout a pharmaceutical product's lifecycle.

Pharma 4.0 – the Key Enabler for Successful Digital Transformation in Pharma
Part 1: Building a Business Case for Pharma 4.0

Relocation of Chemicals Production Footprint in Full Swing
A new Horváth study based on interviews with CxOs of Europe’s top chemical corporations reveals: The majority of board members expects no or only weak growth for the current year.

ECA Foundation Aims to Become Largest Pharma Association for GMP/GDP Compliance
The ECA Foundation, one of the most important not-for-profit organizations for regulatory expertise in the pharmaceutical industry, aims to become the largest independent GMP/GDP organization in the world.

Pharma 4.0—the Key Enabler for Successful Digital Transformation in Pharma
Part 3: Seven Theses for successful Digitalization in Pharma