
Herstellung steriler Arzneimittel
Wie Anlagen fit für den GMP Annex 1 werden
-
 ©Andrii Zastrozhnov - stock.adobe.com
©Andrii Zastrozhnov - stock.adobe.com
Mit dem Annex 1 ist im August 2023 eine umfassende Überarbeitung des EU-GMP-Leitfadens für die Herstellung von sterilen Produkten in Kraft getreten. Es handelt sich um die größte einzelne Änderung der Regulierungsansätze seit Jahrzehnten.
Annex 1 ist ein regulatorischer Schwerpunkt der Europäischen Union, und auch jedes Arzneimittel, das in die EU eingeführt wird, muss dessen Anforderungen erfüllen. Viele internationale und US-amerikanische Organisationen haben zu den Überarbeitungen beigetragen, darunter die FDA, die WHO und die ISPE. Daher kann er als echter globaler Standard betrachtet werden, unter der Erwartung, dass seine bahnbrechende Arbeit von den wichtigsten Regulierungsbehörden übernommen wird.
Als Partner für die Biopharmaindustrie entwickelt Zeta effiziente kundenspezifische Lösungen für die aseptische Prozessführung. Die Vision des Unternehmens: Lebensrettende Medikamente sollen die Patienten schneller erreichen. Natürlich steht die Sicherheit der Patienten immer im Fokus. Deshalb wissen wir aus langjähriger Erfahrung, wie man maximale Produktsicherheit und die Einhaltung der zahlreichen Normen und Richtlinien der Pharmaindustrie sicherstellt.
Kontaminationskontrolle als konsolidierte Gesamtstrategie
Die Änderungen im Annex 1 rücken die Kontaminationskontrolle in den Vordergrund, ganz im Einklang mit der Entwicklung der Industrie in Richtung eines stärker wissensbasierten, risikobezogenen Ansatzes. In Analogie zum anerkannten Quality by Design-Ansatz (QbD) muss auch die Sterilität „durch Design“ und nicht durch Testen des Endprodukts sichergestellt werden. Dies wird durch die Entwicklung von Mechanismen zum Schutz des Produkts vor Kontaminationsquellen durch Mikroorganismen, Partikel und Endotoxine/Pyrogene erreicht.
Ein zentrales Merkmal des QbD-Ansatzes ist die Anwendung von Methoden des Risikomanagements bei der Konzeption, Entwicklung und Herstellung von Arzneimitteln. Das Hauptziel des Qualitäts-Risikomanagements (QRM) besteht natürlich darin, die Sicherheit der Patienten zu gewährleisten. Mit Blick auf den Aspekt der Sterilität erfordert ein solcher Ansatz eine robuste und wissenschaftlich vertretbare Kontaminationskontrollstrategie (Contamination Control Strategy, CCS).
In Annex 1 wird ausdrücklich die Kontaminationskontrolle als konsolidierte Gesamtstrategie gefordert. Die CCS ist ein übergeordnetes Dokument, das sich auf eine Anlage oder einen Prozess innerhalb dieser Anlage bezieht und den Ansatz des Herstellers zur Minimierung der Kontamination in jedem Prozessschritt darstellt. Es erfordert ein umfassendes Verständnis des Prozesses, des Equipments und der Anlage, um Kontaminationsereignisse ganzheitlich zu bewerten und die Festlegung geeigneter Maßnahmen oder CAPAs (Corrective and Preventive Actions) zu ermöglichen, wie sie in den GMP-Richtlinien definiert sind. Das CCS sollte die Gestaltung von Anlagen und Prozessen und die dazugehörige Dokumentation, die Räumlichkeiten und das Equipment und – sehr wichtig – das Personal umfassen, ist aber nicht darauf beschränkt. Das CCS wird als „lebendiges" Dokument betrachtet, das kontinuierlich mit den zunehmend verfügbaren Technologien zur Vermeidung von Kontaminationen aktualisiert wird.
Die Auswirkungen des Annex 1 auf die Sterilherstellung sind Gegenstand vieler Diskussionen in der pharmazeutischen Fachwelt. Kaum eine Überarbeitung hat in den letzten Jahren in der Branche so viel Aufsehen erregt wie der Annex 1, der in 98 Ländern rund um den Globus gelten soll. Natürlich sind die Auslegung und Umsetzung dieses globalen Dokuments durch Behörden und Industrie eine komplexe Angelegenheit.
Unser Wissen in der GMP-konformen Prozessführung und unsere Erfahrung in der Konzeption aseptischer Anlagen erlaubt es uns, Kunden bei der Umsetzung der Annex-1-Richtlinien optimal zu unterstützen, Lücken zu identifizieren und robuste Lösungen zu entwickeln.
Best Practice: Machbarkeitsstudie zum Design einer Point-of-Fill Filtration
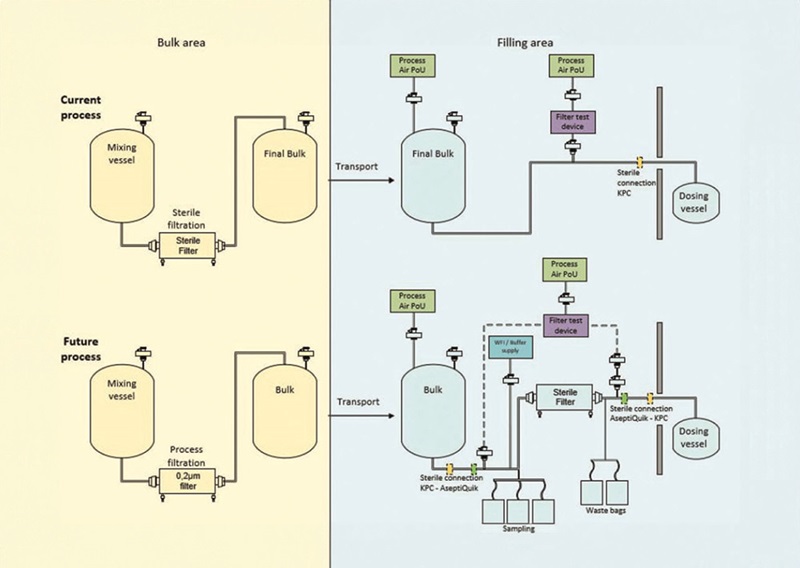
Das Kapitel „Produktion und spezifische Technologien“ des Annex 1 enthält Leitlinien für die Sterilisation von Produkten, Equipment und Verpackungskomponenten. Eine spezifische Anforderung, die in diesem Kapitel (8.79) aufgeführt ist, lautet wie folgt: „Wenn das Produkt nicht in seinem endgültigen Behälter sterilisiert werden kann, sollten Lösungen oder Flüssigkeiten durch Filtration über einen sterilen Sterilfilter sterilisiert und anschließend aseptisch in einen zuvor sterilisierten Behälter gefüllt werden.“ Um dieser Anforderung und den eigenen Ansprüchen an die Prozesssicherheit gerecht zu werden, passt ein renommierter Hersteller therapeutischer Proteine seine Produktionsprozesse entsprechend an. Die Änderungen betreffen vier Abfülllinien in einer Anlage in Deutschland, bei welchen eine Sterilfiltration in der Nähe der Abfüllung eingeführt werden soll. Darüber hinaus umfassen sie die Einführung eines Pre-Use-Post-Sterilization-Integrity-Tests (PUPSIT) für die verwendeten Filter.
Um die Lücke zum EU-GMP-Annex 1 durch die Implementierung von Sterilfiltrationssystemen an den Abfüllanlagen zu schließen, ist eine Vielzahl von Lösungsmöglichkeiten denkbar. Die beste Option in Bezug auf Investitions- und Betriebskosten, Ausfallzeiten, Vorlaufzeit, Komplexität in der Handhabung und andere technische und wirtschaftliche Aspekte zu finden, ist eine anspruchsvolle Aufgabe. In enger Zusammenarbeit mit dem genannten Kunden entwickelten wir in einer Machbarkeitsstudie mehrere mögliche technische Lösungen. So konnte sich der Pharmahersteller für die günstigste Montagelösung entscheiden, die es weiterzuentwickeln galt.
Unser Team untersuchte drei Hauptlösungsoptionen für die Einführung einer Sterilfiltration an der Abfüllstelle. Die erste bestand in einer Modifikation der bestehenden Abfülllinien durch die Installation von Equipment zur Sterilfiltration in den Abfüllmaschinen. Eine weitere Möglichkeit war ein Sterilfiltrationssystem aus Edelstahl – entweder stationär mit Inline-Reinigung oder mobil mit Offline-Autoklavierung. Die dritte Option bestand aus einem Einwegsystem (SU), das entweder vor Ort unter einer Laminar-Flow-Einheit montiert oder als vorgefertigte, gebrauchsfertige Einheit verwendet werden konnte.
Die Alternativen wurden eingehend geprüft, ihre Vor- und Nachteile bewertet und detailliert dargestellt. Darüber hinaus wurden eingehende Bewertungen durchgeführt, die verschiedene Bereiche abdeckten – wie Verfahrenstechnik, Automatisierung, Platzbedarf, Validierung, Qualifizierung, Zeitplan und Kosten. Auf der Grundlage dieser gründlichen Untersuchungen war der Kunde in der Lage, eine fundierte Entscheidung über seine bevorzugte Option zu treffen und entschied sich für die Single Use Assembly-Lösung, die nun weiterentwickelt werden sollte.
Um die Risiken systematisch zu bewerten und zu dokumentieren, untersuchten wir die technischen Details und führten eine Analyse (Failure Mode and Effects Analysis, FMEA) durch. Diese funktionale Risikobewertung lieferte eine solide Grundlage für Entscheidungen über weitere Entwicklungsschritte.
Die wichtigsten Anforderungen an den Point-of-Fill-Sterilfiltrationsprozess wurden als Randbedingungen für die entwickelten Lösungen berücksichtigt. Zu diesen Anforderungen gehört gemäß EU-GMP- Annex 1 ein Inline-Pre-Use-Post-Sterilization-Integrity-Test, der für alle Produktfilter zu implementieren ist. Weitere Überlegungen betrafen die Überwachung kritischer Prozessparameter, die Integritätsprüfung und Überwachung von Spülmedien, sterilen Verbindungen und Prozessluftfiltern sowie die Maximierung der Produktgewinnung. Es wurde ein Prozessflussdiagramm entwickelt, das die wichtigsten Geräte, Anschlusstypen, Instrumente und Reinraumklassen enthält.
Projektumsetzung
Unser Team erarbeitete einen Zeitplan für die Projektumsetzung einschließlich der behördlichen Genehmigung und bewertete die damit verbundenen Kosten. Entsprechend den Wünschen des Kunden wird der zusätzliche Prozessschritt unter Berücksichtigung der Prioritäten der einzelnen Produkte schrittweise umgesetzt. Änderungen vor Ort werden in freie Produktionsslots oder Produktionsstillstände eingeplant. Es wurde ein Automatisierungskonzept entwickelt und ein Gesamtplan für die Projektdurchführung ausgearbeitet, wobei ZETA die Rolle des EPCM (Engineering, Procurement and Construction Management) Partners zugewiesen wurde.
Eine lückenlose Dokumentation der beschriebenen Bewertungen wurde erstellt und soll die weiteren Entscheidungen des Kunden sowie die behördliche Genehmigung der geplanten Änderungen unterstützen. Für den Pharmahersteller ist klar: Die Machbarkeitsstudie ist ein großer Schritt, um fit für die Anforderungen des EU-GMP Annex 1 zu werden!
Autor: Michael Christ, Senior Project Manager, ZETA GmbH, Lieboch, Österreich
-
 Michael Christ, ZETA GmbH © Zeta
Michael Christ, ZETA GmbH © Zeta
„Für die Implementierung von Sterilfiltrationssystemen an den Abfüllanlagen sind viele Lösungsmöglichkeiten denkbar.“










