
Qualifizierung von Reinräumen - Teil 3
Die Qualifizierungsphasen DQ, IQ, OQ, PQ
-
 © Gina Sanders/Fotolia.com
© Gina Sanders/Fotolia.com -
 Prof. Gernod Dittel, Dittel Engineering
Prof. Gernod Dittel, Dittel Engineering -
 Christian Uhl, Dittel Engineering
Christian Uhl, Dittel Engineering -
 Tab. 3: Empfohlene Grenzwerte für die mikrobiologische Überwachung reiner Bereiche im Betriebszustand [14]
Tab. 3: Empfohlene Grenzwerte für die mikrobiologische Überwachung reiner Bereiche im Betriebszustand [14] -
 Tab. 2: Maximal erlaubte Zahl von Partikeln in der Umgebungsluft nach Reinraumklasse [10]
Tab. 2: Maximal erlaubte Zahl von Partikeln in der Umgebungsluft nach Reinraumklasse [10] -
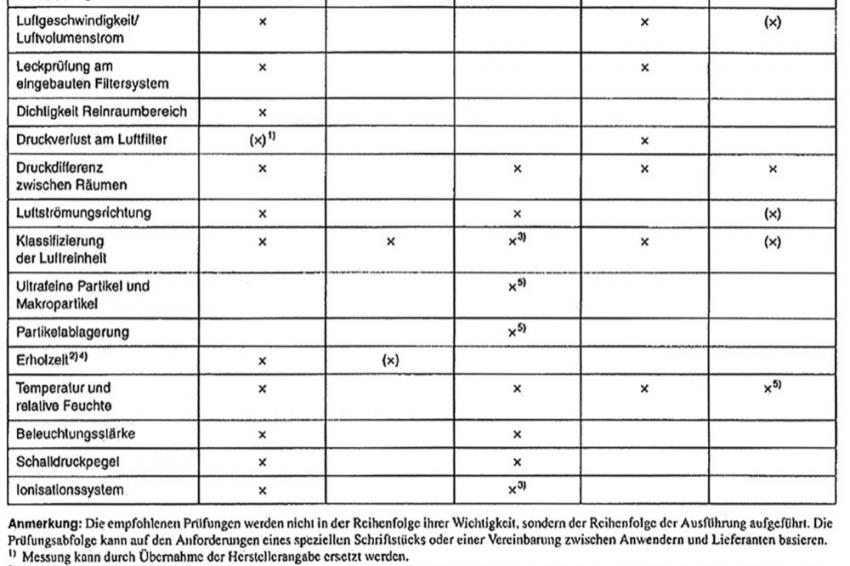 Tab. 1: Empfohlene Prüfungen für die Anlage (Mindestanforderungen) [9]
Tab. 1: Empfohlene Prüfungen für die Anlage (Mindestanforderungen) [9]
ReinRaumTechnik - Die Qualifizierung von Anlagen der Reinraumtechnik ist eine aufwändige Nagelprobe, bei der es für die Betriebe um alles geht. Für viele von ihnen ist der Nachweis der Fähigkeit, die saubere Produktion im Reinraum tatsächlich zu beherrschen, keine freiwillige Kür, sondern notwendige Pflicht.
In der Pharmabranche ist die Qualifizierung die Voraussetzung dafür, dass überhaupt produziert werden darf. Mit dem regulären Betrieb darf nämlich erst begonnen werden, nachdem die Geeignetheit der Räume festgestellt wurde. Mit anderen Worten: nach der „Qualifizierung". Im ReinRaum Tutorial „Qualifizierung von Reinräumen" erläutert Prof. Gernod Dittel die Richtlinien und Maßnahmen zur Qualifizierung. Teil 3 beschäftigt sich mit den Qualifizierungsphasen DQ, IQ, OQ, PQ und im finalen Teil 4 folgen in der kommenden Ausgabe die Themen Änderungskontrolle, Requalifizierung und Abnahmeinspektion.
Die Designqualifizierung (DQ)
Nach dem QMP als Basisdokument ist die Designqualifizierung (DQ) der erste Schritt der Prüfung, ob die Reinraumtechnische Anlage dem EU-GMP-Leitfaden entspricht.[1] Zum Prüfumfang der DQ gehören die Reinraumhülle (Wand, Decke, Boden mit jeweiligen Einbauten), die Lüftungsanlage und Kanäle mit Einbauten, die Steuer- und Regelungstechnik der Lüftungsanlage sowie alle im QMP definierten zu qualifizierenden Systeme. Neben den GMP-Anforderungen wird in der DQ auch geprüft, ob die Anlage weiteren Normen wie der EN ISO 14644 und der VDI 2083 sowie den Kundenspezifikationen (User Requirement Specifications - URS) entsprechen. Alle daraus resultierenden Anforderungen sollten im Zuge des DQ-Plans in einer Prüfliste zusammengefasst werden. Anhand dieser Liste werden die Planungsunterlagen geprüft.
Weiterer Bestandteil der DQ ist eine Risikoanalyse, in der das komplette System auf mögliche Fehler oder Schwachstellen hin betrachtet wird. Mit der Abarbeitung der Prüfliste und der Bewertung der Anlage durch die Risikoanalyse wird im DQ-Bericht eindeutig Stellung zur geplanten Anlage genommen. Die frühzeitige Prüfung der Planungsunterlagen durch die DQ hilft, Mängel in der Planung zu erkennen. Würden Planungsmängel erst in späteren Projektphasen wie bei Baubeginn, Inbetriebnahme oder Abnahme erkannt, würde ihre Behebung wesentlich mehr Ressourcen und Kosten in Anspruch nehmen.
Die Installationsqualifizierung (IQ)
Mit der Installationsqualifizierung (IQ) wird sichergestellt und dokumentiert, dass die Anlage so installiert wurde, wie es die normativen und benutzerspezifischen Anforderungen verlangen. [2] Die IQ wird nach Aufbau der Anlage durchgeführt, meist nach der technischen Abnahme. Es ist sinnvoll, die IQ in zwei Schritte aufzuteilen. Im ersten Schritt wird die vom Lieferanten erstellte Bestandsdokumentation mit der in der DQ geprüften Planungsdokumentation verglichen, um zu prüfen, ob die installierte Anlage gemäß der Planung und somit gemäß den Anforderungen gebaut wurde. Des Weiteren wird in diesem Schritt die Bestandsdokumentation auf Vollständigkeit und Fehlerfreiheit überprüft. Im zweiten Schritt wird dann untersucht, ob die Anlage selbst entsprechend der Bestandsdokumentation gebaut wurde und die Ausführung dem EU-GMP-Leitfaden entspricht. Um Abweichungen zu beurteilen, kann unter Umständen eine Risikoanalyse vonnöten sein.
Anforderungen an Produktionsräume, die bei der IQ geprüft werden, sind zum Beispiel:
- Innen liegende Oberflächen sollten glatt, frei von Rissen, Beschädigungen und offenen Fugen sein.
- Oberflächen sollten keine Partikel emittieren, leicht zu reinigen und zu desinfizieren sowie beständig gegen Reinigungs- und Desinfektionsmittel sein. [3]
- Das Lüftungssystem sollte das Produkt und die Prozesse nicht negativ beeinflussen. [4]
- Das Lüftungssystem sollte keine Partikel erzeugen, die Partikelkonzentration wirksam verdünnen, beziehungsweise die Partikel verdrängen.
Die Funktionsqualifizierung (OQ)
Die Funktionsqualifizierung (OQ) soll dokumentieren, dass die Anlage und ihre Bestandteile im Rahmen der vorgesehenen Betriebsbereiche den Erwartungen gemäß funktionieren. [5] Zu diesem Zweck sollten im Zuge des OQ-Plans Prüflisten für die Funktionsprüfung erstellt werden. Prüfpunkte sind zum Beispiel Funktions- und Sicherheitsprüfungen der Lüftungsanlage und im Reinraum (zum Beispiel der Schleusensteuerung und Zugangskontrollen) sowie messtechnische Funktionsprüfungen im Zustand „at rest".
Für die messtechnischen Prüfungen definiert der EU-GMP-Leitfaden die zwei Zustände „at rest" (Ruhezustand) und „in operation" (Betriebszustand). Der Zustand „at rest" ist „der Zustand, in dem die Produktionsausrüstung installiert und ohne Anwesenheit des Bedienpersonals in Betrieb ist" [6]. Demgegenüber bedeutet „in operation" den Zustand, „in dem die Anlage in der vorhergesehenen Art mit der festgelegten Personenzahl betrieben wird" [7]. In Tabelle 1 sind die von der VDI-Richtlinie 2083 Blatt 3 empfohlenen Prüfungen aufgelistet. Die abgebildeten Messungen im Zustand „as built" werden in der Praxis häufig mit dem Zustand „at rest" zusammengefasst.
Für die Messung zur Klassifizierung der Luftreinheit macht der EU-GMP-Leitfaden gemäß der jeweiligen Reinraumklasse A bis D spezifische Vorgaben (Tab. 2). Bei den messtechnischen Prüfungen soll „nur geschultes Personal eingesetzt" [8] und es dürfen nur kalibrierte Messsysteme verwendet werden. Wie in den vorherigen Qualifizierungsphasen kann es auch in der OQ nötig sein, gewisse Abweichungen und Mängel in eine Risikoanalyse zu überführen und dort zu bewerten.
Die Leistungsqualifizierung (PQ)
Bei der Leistungsqualifizierung (PQ) verifiziert der Anwender, ob die Anlage und ihre Teile, „so wie sie miteinander verbunden wurden, auf der Grundlage der genehmigten Prozessmethode und Produktspezifikation effektiv und reproduzierbar funktionieren"[11]. Verlangt werden Tests mit Produktionsmaterialien, geeigneten Ersatzmaterialien oder simulierten Produkten. [12] Die PQ einer Reinraumtechnischen Anlage beschränkt sich in der Regel auf die Messungen im Zustand „in operation". Dabei wird überprüft, ob die Anlage auch unter Volllast, also mit der vorgesehenen Anzahl an Personal und bei laufenden Produktionsanlagen, die spezifizierten Parameter einhält. Neben den in Tabelle 1 aufgeführten Messungen müssen auch mikrobiologische Nachweismessungen erfolgen (vgl. Grenzwerte in Tabelle 3). Die Ergebnisse der PQ werden wie bei allen vorherigen Qualifizierungsphasen in einem Bericht festgehalten. Erst nach einer erfolgreichen Leistungsqualifizierung der Reinraumtechnischen Anlage kann mit der Prozessvalidierung begonnen werden. [13]
Der Qualifizierungsabschlussbericht
Sind über die Qualifizierungsphasen hinweg alle definierten Maßnahmen erledigt, kann der Anwender die Qualifizierung der Reinraumtechnischen Anlage mit einem Qualifizierungsabschlussbericht abschließen. Darin fasst er die Ergebnisse aller Qualifizierungsphasen (DQ, IQ, OQ, PQ) zusammen. Alle Änderungen (Change Control) werden kurz begründet. Kritische Mängel müssen vor Freigabe des Qualifizierungsabschlussberichts behoben sein, unkritische Mängel können später abgearbeitet werden. Der Betrieb hat jedoch darauf zu achten, dass die Mängelbeseitigung auch nach der Freigabe fortgeführt wird.[15] Des Weiteren sollte der Qualifizierungsabschlussbericht einen Dokumentennachweis mit allen während der Qualifizierung erstellten Dokumenten beinhalten und den Qualifizierungsstatus der Reinraumtechnischen Anlage eindeutig kennzeichnen.
Literatur
[1] Ebenda: Nr. 9
[2] EU-GMP-Leitfaden Anhang 15 (2001): Qualifizierung und Validierung, Glossar
[3] Aide-Mémoire 07121105: Inspektion von Qualifizierung und Validierung in pharmazeutischer Herstellung und Qualitätskontrolle, Nr. 4.1.2
[14] Ebenda
[5] EU-GMP-Leitfaden Anhang 15 (2001): Qualifizierung und Validierung, Glossar.
[6] EU-GMP-Leitfaden Anhang 1 (2009): Herstellung steriler Arzneimittel, Nr. 3.
[7] Ebenda
[8] Ebenda: Messtechnik in der Reinraumluft, Nr. 4.1
[9] VDI-Richtlinie 2083 Blatt 3 (2005): Messtechnik in der Reinraumluft, Nr. 4.1.1.2
[10] EU-GMP-Leitfaden Anhang 1 (2009): Herstellung steriler Arzneimittel, Nr. 4
[11] EU-GMP-Leitfaden Anhang 15 (2001): Qualifizierung und Validierung, Glossar
[12] Ebenda: Nr. 17a
[13]Hiob, Michael (2010): Leistungsqualifizierung, in: Maas, Anita / Peither, Barbara / Peither, Thomas (Hg.): GMP-Berater. Nachschlagewerk für Pharmaindustrie und Lieferanten, Schopfheim, Maas&Peither AG - GMP-Verlag, Nr. 6.A.6
[14] EU-GMP-Leitfaden Anhang 1 (2009): Herstellung steriler Arzneimittel, Nr. 19
[15] Reuter, Ulrike (2010): Qualifizierungsbericht, in: Maas, Anita / Peither, Barbara / Peither, Thomas (Hg.): GMP-Berater. Nachschlagewerk für Pharmaindustrie und Lieferanten, Schopfheim, Maas&Peither AG - GMP-Verlag, Nr. 6.C.3
Kontakt
Dittel Engineering
Von-Velsen-Str. 8
82431 Kochel
Deutschland











