
Better Regulation of Pharmaceuticals
Towards a Simpler, Clearer and More Flexible Framework 0n Variations
-
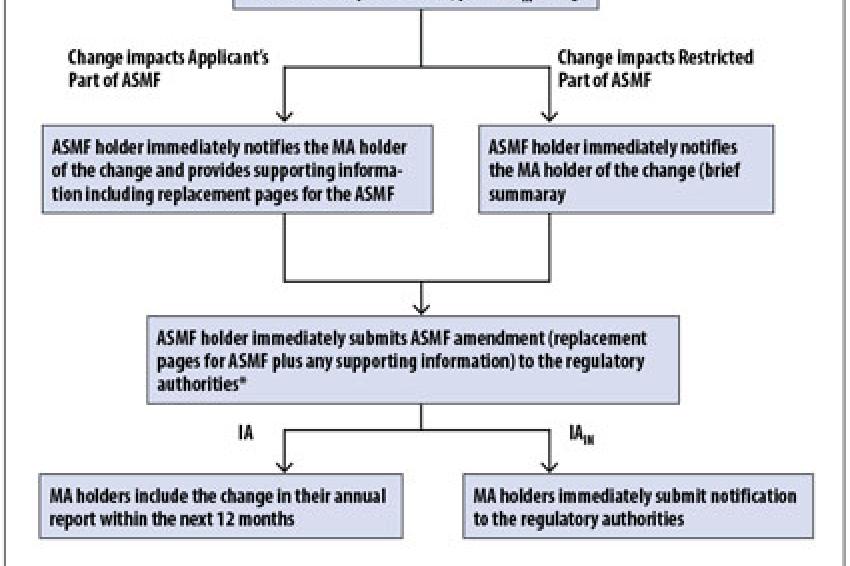 Fig. 1: Proposal for Handling Type IA / IAIN Changes to Active Substance Master Files
Fig. 1: Proposal for Handling Type IA / IAIN Changes to Active Substance Master Files
Medicines are regulated throughout their lifetime. All changes subsequent to their placement on the EU market, for example, changes in the production process, the packaging, the address of the manufacturer, are considered in legal terms as "variations," and must be handled according to a complex legislative framework: the "Variations Regulation."
While regulating variations is essential to ensure that medicines remain safe and effective, prior to the revision of the Variations Regulation, a large majority of products fell outside the scope of community rules and were subject to divergent national rules. This lack of harmonization had negative consequences in terms of administrative burden, both for industry and national regulatory authorities, and in the overall functioning of the internal market.
The objective of the revision of the Variations Regulation, initiated by the EU Commission, Enterprise and Industry, was to simplify the legislative framework governing variations. The new Regulation (1234/2008) applies to variations as of Jan. 1, 2010, as per the current scope of the regulation, namely marketing authorisations (MAs) obtained via the centralised, decentralised (DCP) and mutual recognition (MRP) procedures.
Broadening of Regulation Scope
The broadening of the scope of the regulation to include national submissions can be exercised by the EU Commission from January 2011. This will bring a major benefit to industry since the majority of MAs are currently still purely national. In particular, the fixed approval timelines will be a significant benefit since these will allow implementation of the variation within a predictable timeframe.
EU Commission Recognizes That Change is Vital
Change is vital to enable innovation and continual improvement of pharmaceutical products and manufacturing processes, to increase quality and safety, reduce environmental impact and accommodate the evolution of science and technology. The manufacture of APIs is no exception to this. Change is the most important tool for industry to create sustainable growth and to remain competitive in continuously changing circumstances.
In order to make changes, the dedicated API industry must work closely with the MA holder. Not only must the impact of the change to the API on the medicinal product be assessed, it is the MA holder who must submit the variation application. For the MA holder, this means the filing of variations in different countries, for different formulations, via different procedures with different approval times. It may be that full approval for implementation is only obtained after several or even many years. Under these difficult circumstances, it is easy to understand why MA holders are often reluctant to support changes proposed by their API suppliers. This may lead to the change being blocked completely. For many API manufacturers, making a change under the old system was virtually impossible. It is not surprising then that the Commission's initiative towards a simpler, clearer and more flexible framework on variations was very much welcomed by the API industry.
One of the changes in the new regulation is the introduction of a "do and tell" procedure for Type IA variations. Some Type IA variations will require immediate notification after implementation (denoted by the subscript "IN"); the remainder should be notified within 12 months following implementation. Clearly, this will reduce the regulatory burden for the MA holder, however, it is imperative that the active substance master file (ASMF) holder also benefits. While it is acknowledged that the regulation does not allow for the ASMF holder themselves to submit an "annual report," direct contact between the authorities and the ASMF holder is essential in order to keep the ASMF up-to-date and current. This is required because in a multi-customer/multi-authority environment, a new or existing customer may submit a new MA application at any time and for MRP / DCP, it is essential that all authorities have the same version of the ASMF. The direct contact is also needed in order for the ASMF holder to fulfil his responsibilities in accordance with the commitment made in the Letter of Access (CPMP/QWP/227/02 Rev 1 Annex 2).
The Active Pharmaceutical Ingredients Committee (APIC) therefore, proposes that the ASMF holder informs both the MA holder and the authorities of a Type IA change at the time of implementation. The MA holder would subsequently include the variation in his annual report at his discretion (fig. 1).
Regular Update of the Classification of the Changes
Another change in the new regulation is the omission of the detailed annex containing the classification of Type IA and IB variations. Under the old regulations, any variation that was not listed as Type IA or IB (and was not an extension) was Type II by default. Now, in order to facilitate regular revision, the classifications are listed in a guideline rather than in the regulation itself and any variation that is not listed as Type IA or Type II (and is not an extension) is Type IB by default. (Examples of Type IB variations are provided but the list is not exhaustive.) This default principle should reduce the number of changes blocked since submission and approval times for API changes will interfere less with medicinal product changes or clinical changes etc.
In conclusion, the EU Commission initiative for better regulation of pharmaceuticals is a significant step forward in harmonizing the handling of variations in the whole of the European Union and is now reaching out to the EU health authorities and industry to put this into practise in terms of timelines, documentation requirements and to engage in employing the new principles laid out in regulation 1234/2008 and its two supporting guidelines to its full intent.
Hilde Vanneste



