
GDUFA’s Impact on the API Industry
Are Fees Prohibitive or the Price of Doing Business?
-
 Time will tell if DMF fees are prohibitive to API suppliers or if these payments are simply accepted as the price of doing business. © Rrraum - Fotolia.com
Time will tell if DMF fees are prohibitive to API suppliers or if these payments are simply accepted as the price of doing business. © Rrraum - Fotolia.com -
 Michael Glessner, Pharmaceutical Research Analyst, Thomson Reuters
Michael Glessner, Pharmaceutical Research Analyst, Thomson Reuters -
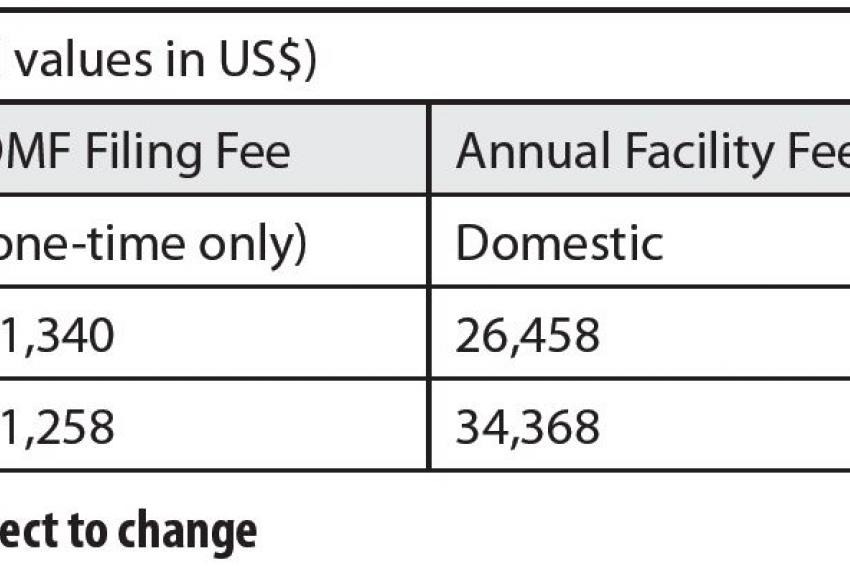 Fig. 1: GDUFA Fee structure.
Fig. 1: GDUFA Fee structure. -
 Fig. 2: US Type II Active DMFs.
Fig. 2: US Type II Active DMFs.
One-Time Fee - On Oct. 1, 2012, the U. S. Food and Drug Administration (FDA) implemented the long-discussed Generic Drug User Fee Act (GDUFA) , thereby authorizing the agency to collect fees from any manufacturers worldwide who wish for their active pharmaceutical ingredients (APIs) to be eligible for use in generic drugs sold in the U. S. According to the legislation, a one-time fee is assessed the first time a Type II Drug Master File (DMF) is referenced in an Abbreviated New Drug Application (ANDA).
In addition, manufacturers must pay annual facility fees for each location producing at least one generic API for use in the U.S. market (fig. 1).
With the additional revenue, targeted to be $299 million annually, the FDA plans to add resources in the hopes of accomplishing two important goals. First, they aim to cut into the large backlog of ANDAs awaiting review and to reduce average review time moving forward. Second, the FDA wants to increase the breadth and frequency of API manufacturer facility inspections, particularly in less regulated markets where previous inspections by any governing agency may have been non-existent.
Despite the additional fees that will be incurred, many API manufacturers are supportive of the GDUFA legislation. Reduced ANDA review time will help some generics reach the market (and start generating revenue) more quickly. Manufacturers in regulated markets who were already subject to regular inspections by the FDA or the European Medicines Agency (EMA) are eager for their low-cost competitors in less regulated markets to be exposed to a similar level of scrutiny. Some in the industry believe that the FDA's Available for Reference List of DMFs with paid fees (available for download on the FDA website) will help to distinguish legitimate API producers from companies who use regulatory filings to market products they may or may not have the capability to produce.
GDUFA Effects on the API Industry
One possible result of the GDUFA legislation could be a change in the growing trend of DMF filings originating from less-regulated markets. While a significant portion of APIs in the U.S. market continues to come from Italy and Spain, we have seen a sizable increase in the market share held by Indian companies. According to the Chemical Pharmaceutical Generic Association, the market share held by Indian companies quadrupled between 2005 and 2010.
Figure two, created with data from Thomson Reuters Newport Premium shows the total number of currently active U.S. DMFs filed each year since 2000 and breaks down those submissions by the country where DMF manufacturers' corporate headquarters are located. Total DMF count is not a direct measure of regional API production or presence in the U.S. market; many DMFs are filed but never referenced, and some companies can avoid filing DMFs altogether by placing pertinent information directly in their ANDA submission rather than submitting it as a separate document. DMF filings do, however, provide a strong indication of which API manufacturers have an interest in penetrating the U.S. drug market; it is clear from this chart that over the past decade more and more of that interest has come from companies based in India and China.
While we expect continued growth in the number of DMF filings from India and China, the rate at which this growth occurs compared to regulated markets may be curbed in response to the GDUFA legislation. Filing fees as well as the challenges of preparing for and responding to FDA inspections may serve to close the price gap between regulated and unregulated suppliers and slow the prolific rate of Indian and Chinese DMF filings.
Small companies who manufacture only a handful of APIs will find it more difficult to recoup the annual facility fee than large companies who can spread the cost across many products. Smaller players may choose to merge with other small companies, work with a purchaser willing to share costs or decide that the new fees are a prohibitive barrier to U.S. market participation.
Companies with multiple production facilities may decide to consolidate production of APIs bound for the U.S. in an attempt to avoid multiple facility fees. Potent, low-dose APIs that take up less production space could be a better return on investment than a large volume API that limits a company's ability to manufacture multiple products in one facility.
Supplier-Customer Relationship
One interesting sub-plot to the GDUFA story will be the effect of this legislation on the relationship between API suppliers and their customers. Suppliers who make an API exclusively for a customer may look for the customer to cover the cost of the DMF filing fee. Other suppliers may look to line up multiple customers before allowing their DMF to be referenced. API producers who make their living as a "secondary source," supplying generic drug companies with API only in times of shortage or primary supply chain breakdown, could find themselves paying DMF fees for APIs they never sell. These companies may seek some financial incentive from the purchasers who are referencing their DMFs.
Customers, in turn, may take extra care to find a reputable API supplier because if an initial supply agreement falls apart, in addition to paying the new supplier for cost development, validation and stability there is currently a $25,760 fee associated with the submittal of the Prior Approval Supplement (PAS) necessary to change API suppliers. This may drive business back toward API suppliers in more regulated regions like the U.S. and Europe.
Moving Forward
With the GDUFA program still in its infancy, it is difficult to predict not only how the legislation will change the industry, but how long it will take before these changes unfold. Many skeptics believe that the FDA goal of reviewing 90% of ANDAs within 10 months of submission by the end of the program's fifth year is unrealistic. Even with the new revenue, the prospect of hiring and training new employees, addressing the ANDA backlog and cutting two years off the current average review time seems a daunting task. The FDA has announced that, while it will continue to target annual revenue of $299 million from the GDUFA program, DMF filing fees and annual facility fees will increase during FY 2014-2017 (fig. 1). Time will tell if those fees prove prohibitive to API suppliers or if these payments are simply accepted as the price of doing business.
Contact
Thomson Reuters
215 Commercial Str.
Portland, Maine 04101
+1 207 8719700
+1 207 8719800



